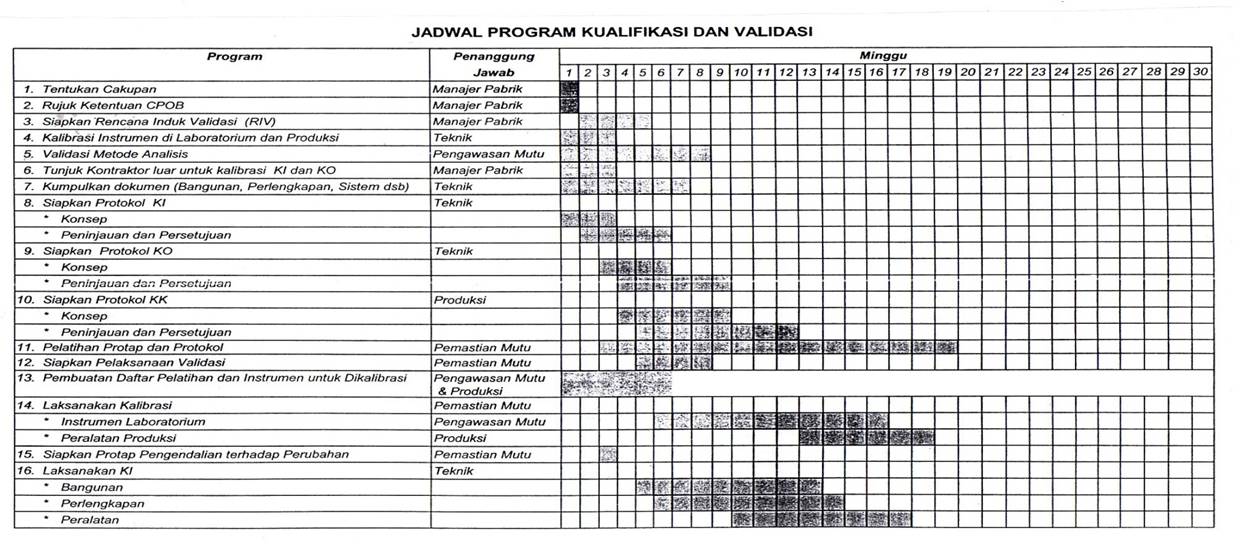
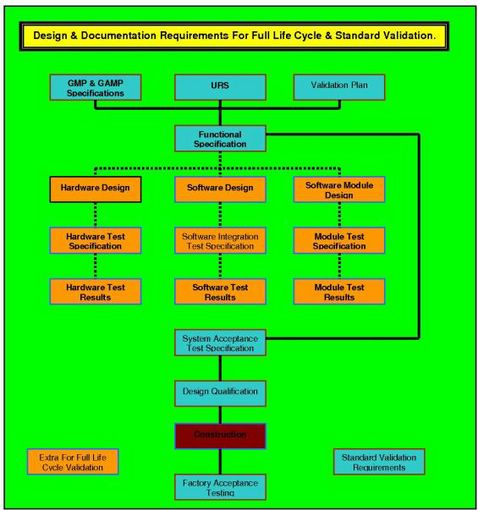
Validation (drug manufacture)
From Wikipedia, the free encyclopedia
History
The concept of validation was first proposed by two
Food and Drug Administration (FDA) officials, Ted Byers and Bud Loftus, in the mid 1970’s in order to improve the quality of pharmaceuticals (Agalloco 1995). It was proposed in direct response to several problems in the sterility of large volume
parenteral market. The first validation activities were focused on the processes involved in making these products, but quickly spread to associated processes including environmental control, media fill, equipment
sanitisation and purified water production. In a guideline on process validation the FDA define (FDA 1987) Validation as: "Establishing documented evidence that provides a high degree of assurance that a specific process will consistently produce a product meeting its pre-determined specifications and quality attributes."
Computer System ValidationThis requirement has naturally expanded to encompass computer systems used both in the development and production of, and as a part of
pharmaceutical products and
medical devices, as computer systems have entered more and more into the main stream of drug and medical device production. In 1983 the FDA published a guide to the inspection of Computerised Systems in Pharmaceutical Processing, also known as the ‘bluebook’ (FDA 1983). Recently both the American FDA and the UK MHRA have added sections to the regulations specifically for the use of computer systems, for the MHRA this is Annex 11 of the EU GMP regulations (EMEA 1998), and the FDA introduced 21 CFR Part 11 for rules on the use of electronic records, electronic signatures (FDA 1997). The FDA regulation is harmonized with ISO 8402:1994 (ISO 1994), which treats “
verification” and “validation” as separate and distinct terms. On the other hand, many software engineering journal articles and textbooks use the terms "verification" and "validation" interchangeably, or in some cases refer to software "
verification, validation, and
testing (
VV&T)" as if it is a single concept, with no distinction among the three terms. The General Principles of Software Validation (FDA 2002) defines verification as “Software verification provides objective evidence that the design outputs of a particular phase of the software development life cycle meet all of the specified requirements for that phase.” It also defines Validation as “Confirmation by examination and provision of objective evidence that software specifications conform to user needs and intended uses, and that the particular requirements implemented through software can be consistently fulfilled”
Goal of Validation
The goal for the regulators is to ensure that quality is built into the system at every step, and not just tested for at the end, as such validation activities will commonly include training on production material and operating procedures, training of people involved and monitoring of the system whilst in production. In general, an entire process is validated; a particular object within that process is verified. The regulations also set out an expectation that the different parts of the production process are well defined and controlled, such that the results of that production will not substantially change over time. This also extends to include the development and implementation as well as the use and maintenance of computer systems. The software validation guideline states: “The software development process should be sufficiently well planned, controlled, and documented to detect and correct unexpected results from software changes.”
Why ValidateThe concept of validation was first developed for equipment and processes and derived from the engineering practices used in delivery of large pieces of equipment that would be manufactured, tested, delivered and accepted according to a contract (Hoffmann et al. 1998). The use of validation spread to other areas of industry after several large-scale problems highlighted the potential risks in the design of products. The most notable is the
Therac-25 incident, (Leveson & Turner 1993). Here, the software for a large radiotherapy device was poorly designed and tested In use, several interconnected problems lead to several devices giving doses of radiation several thousands of times higher than intended, which resulted in the death of three patients and several more being permanently injured. Weichel (2004) recently found that over twenty warning letters issued by the FDA to pharmaceutical companies specifically cited problems in Computer System Validation between 1997 and 2001. Validation is intended to provide assurance of the quality of a system or process through a quality methodology for the design, manufacture and use of that system or process, that cannot be found by simple testing alone (McDowall 2005).
Basics
In general the validation process consists of three major phases:
Installation Qualification (IQ) - Demonstrate that the process or equipment to be qualified meets all specifications, is installed correctly, and all required components and documentation needed for continued operation are installed and in place.
Operational Qualification (OQ) - Demonstrate that all facets of the process or equipment are operating correctly.
Performance Qualification (PQ) - Demonstrate that the process or equipment performs as intended in a consistent manner over time.
Although IQ/OQ/PQ are the main phases of the entire validation process, some definitions include other phases such as Design Qualification (DQ).
Scope of Computer ValidationThe definition of validation above discusses production of evidence that a system will meet its specification. This definition does not refer to a computer application or a computer system but to a process. The main implications in this are that validation should cover all aspects of the process including the application, any hardware that the application uses, any interfaces to other systems, the users, training and documentation as well as the management of the system and the validation itself after the system is put into use. The PIC/S guideline (PIC/S 2004) defines this as a ‘computer related system’ Much effort is expended within the industry upon validation activities, and several journals are dedicated to both the process and methodology around validation, and the science behind it (Smith 2001;Tracy & Nash 2002;Lucas 2003;Balogh & Corbin 2005).
Risk Based Approach for Computer Systems
In recent years, a risk-based approach has been adopted within the industry, where the testing of computer systems (emphasis on finding problems) is wide-ranging and documented but not heavily evidenced (e.g. 100's of screen prints are not gathered during testing). The subsequent validation or verification of computer systems targets only the "GxP critical" requirements of computer systems, and in this case evidence (e.g. screen prints) is gathered to document the validation exercise. In this way it is assured that systems are thoroughly tested, and that validation and documentation of the "GxP critical" aspects is performed in a risk-based manner optimising effort and ensuring that computer system's fitness for purpose is demonstrated.
The overall risk posed by a computer system is now generally considered to be a function of system complexity, patient/product impact, and pedigree (Configurable-Of-The-Shelf or Custom-written for a certain purpose). A lower risk system should merit a less in-depth specification/testing/validation approach. (e.g. The documentation surrounding a spreadsheet containing a simple but "GxP" critical calculation should not match that of a Chromatography Data System with 20 Instruments)
Determination of a "GxP critical" requirement for a computer system is subjective, and the definition needs to be tailored to the organisation involved. However in general a "GxP" requirement may be considered to be a requirement which leads to the development/configuration of a computer function which has a direct impact on patient safety, the pharmaceutical product being processed, or has been developed/configured to meet a regulatory requirement. In addition if a function has an direct impact on GxP data (security or integrity) it may be considered "GxP critical".
Problems in ValidationMany practitioners within pharmaceutical validation have commented on the increasing requirement for documentation and testing, which does not give extra assurance of the safety or quality of the product. Akers (1993) said: “QA and Regulatory Affairs departments within industry and Regulatory Agencies are obstacles to reduced testing since each group has their own interests to protect. Clearly, reduced auditing and testing and therefore reduced staffing is an unwelcome notion to many middle managers.” And “From the FDA perspective, it has always been safer to have more tests even if they provide no additional statistical confidence in product safety. The result of this has been that even those creative validation people who could have made validation a real process control tool have been thwarted by other special interest groups.” Powell-Evans (1998) said that “…in its Quasi-form, validation is expensive, inefficient, ineffective and awkward. It hinders progress and clogs up otherwise creditable systems for good drug production.
GMP now, as we all know, should stand for ‘great mounds of paper’. These mounds are produced in a desperate attempt to ensure that every nook and cranny, every nut and bolt, and every roll and shake of an operators lab coat is signed, sealed and delivered to the regulators cold eye. It should be asked whether all this is necessary and / or beneficial, and whether a better method can be found? Or maybe we should try and understand how validation really works, moreover applied, maybe then we can move away from the Quasi approach industry has adopted, towards the view of those in the know...validation works...if you do it right!!” This has led some practitioners to search for better ways to perform development and validation. This includes moves towards measures of Cost of Quality and Risk Assesement to provide systems that perform the job with enough assurance of quality and safety without the burden of un-necessary documentation and planning (Garston Smith 2001).
Problems of Self RegulationIn general, the regulatory agencies, through laws and guidelines provide a broad overview of what they want pharmaceutical companies to provide, but not how to do it. They will
audit the system and list any areas where they feel the approach is not satisfactory, but generally do not provide help on how to fix the situation. This often leads to companies performing more work than is necessary ‘just in case’ or doing less than what is necessary. It is critical that the company has a good understanding of the equipment and the intended use so that the right amount of validation is performed.
Problems in Testing
Testing, metrology, and documentation requirements for validation are challenging. For even relatively simple
computer programs, it quickly becomes impossible to test every permutation and route through the program. This was described by Boehm (1970).
Changing TerminologyGiven the wide range of the pharmaceutical industry from Research and development to Production, Delivery and Sales and the different regulations like GMP and GLP enacted in different ways in different countries the basic terminology used can be different. This may be solved one day by the International Conferences on Harmonisation (ICH) but until then will be a major problem.
See also
GxPGood Manufacturing Practice (GMP)
Good Automated Manufacturing Practice (GAMP)
Verification and ValidationPharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation SchemeRegulation of therapeutic goodsUnited States PharmacopeiaIT Pharma Validation Europe Organization: Free Downloads and InformationReferences
Agalloco, J. (1995), 'Validation: an unconventional review and reinvention', PDA J Pharm Sci Technol., vol. 49, no. 4, pp. 175-179.
Akers, J. (1993), 'Simplifiying and improving Process Validation', Journal of Parenteral Science and Technology, vol. 47, no. 6, pp. 281-284.
ASTM E2537 Guide for Application of Continuous Quality Verification for Pharmaceutical and Biopharmaceutical Manufacturing
Balogh, M. & Corbin, V. (2005), 'Taming the Regulatory Beast: Regulation vs Functionalism', Pharmaceutical Technology Europe, vol. 17, no. 3, pp. 55-58.
Boehm, B. W. (1970), Some information processing implications of air force missions 1970-1980, The Rand Corporation, Santa Monica
EMEA (1998), EUDRALEX Volume 4 - Medicinal Products for Human and Veterinary Use : Good Manufacturing Practice, European Medicines Agency, London
FDA (1983), Guide to Inspection of Computerised Systems (The Blue Book), US Food and Drug Administration, Maryland, USA.
FDA (1987), Guideline on general principles of Process Validation, US Food and Drug Administration, Maryland, USA
FDA (2002), General Principles of Software Validation; Final Guidance for Industry and FDA Staff, US Food and Drug Administration, Maryland, USA
FDA (2004), Part 11: Electronic Records; Electronic Signatures,Code of Federal Regulations, Title 21 - Food and Drugs, Chapter I - Food and Drug administration, Office of the Federal Register, Maryland, USA
Garston Smith, H. (2001), 'Considerations for Improving Software Validation', Journal of Validation Technology, vol. 7, no. 2, pp. 150-157.
Hoffmann, A., Kahny-Simonius, J., Plattner, M., Schmidli-Vckovski, V., & Kronseder, C. (1998), 'Computer system validation: An overview of official requirements and standards', Pharmaceutica Acta Helvetiae, vol. 72, no. 6, pp. 317-325.
ISO (1994), ISO 8402:1994: Quality management and quality assurance -- Vocabulary, International Organization for Standardization, Geneva, Switzerland
Leveson, N. G. & Turner, C. S. (1993), 'An investigation of the Therac-25 accidents', Computer, vol. 26, no. 7, pp. 18-41.
Lucas, I. (2003), 'Testing Times in Computer Validation', Journal of Validation Technology, vol. 9, no. 2, pp. 153-161.
McDowall, R. D. (2005), 'Effective and practical risk management options for computerised system validation', The Quality Assurance Journal, vol. 9, no. 3, pp. 196-227.
PIC/S (2004), Good Practices for Computerised Systems in Regulated "GXP" Environments, Report PI 011-2, Pharmaceutical Inspection Convention, Geneva
Powell-Evans, K. (1998), 'Streamlining Validation', Pharmaceutical Technology Europe, vol. 10, no. 12, pp. 48-52.
Smith, H. G. (2001), 'Considerations for Improving Software Validation, Securing better assurance for less cost', Journal of Validation Technology, vol. 7, no. 2, pp. 150-157.
Tracy, D. S. & Nash, R. A. (2002), 'A Validation Approach for Laboratory Information Management Systems', Journal of Validation Technology, vol. 9, no. 1, pp. 6-14.
Weichel, P. (2004), 'Survey of Published FDA Warning Letters with Comment on Part 11 (21 CFR Part 11)', Journal of Validation Technology, vol. 11, no. 1, pp. 62-66.
Retrieved from "
http://en.wikipedia.org/wiki/Validation_(drug_manufacture)"